lunes, 21 de noviembre de 2016
Causas
Normalmente el factor FGFR3 tiene efecto regulador en el crecimiento de los huesos. En la acondroplasia el receptor de este factor se encuentra mutado, por lo que este se encuentra constitutivamente activo lo cual lleva al acortamiento de los huesos. Las personas con acondroplasia tienen una copia normal del gen del factor FGFR3, pero también tienen una copia mutada. Dos copias del gen mutado es fatal desde antes del nacimiento. En cuanto a la herencia genética, una persona con acondroplasia tiene el 50% de probabilidades de heredar esta enfermedad a sus hijos, lo cual significa que hay un 50% de probabilidades de que cada niño herede esta enfermedad.Por otro lado si ambos padres tienen acondroplasia, sus hijos tienen un 25% de probabilidades de morir poco tiempo después de su nacimiento, y un 50% de probabilidades de que tenga acondroplasia y un 25% de que el niño presente el fenotipo. No todas las personas que nacen con acondroplasia tienen padres con esta misma condición, ya que esto puede ser resultado de una nueva mutación.8 Se cree que esta enfermedad no se adquiere necesariamente por herencia genética, ya que existen nuevas mutaciones de los genes que pueden llevar a una acondroplasia, y esto está asociado principalmente con una edad avanzada de los padres9 (generalmente los mayores a 35 años). Estudios actuales han demostrado que las nuevas mutaciones de los genes para acondroplasia son heredados exclusivamente del padre y que ocurre durante las espermatogénesis; (sobra)pues durante la ovogénesis existe algún tipo de mecanismo regulador que impide la mutación de los genes, sin embargo las mujeres siguen siendo capaces de presentar el fenotipo y genotipo, y (por lo tanto)de transmitir el alelo mutante. Más del 99% de (la) acondroplasia es causada por dos mutaciones diferentes del factor FGFR3. En aproximadamente el 98% de los casos, un punto mutado G a uno A dentro del nucleótido 1138 del gen factor FGFR3 causa una substitución de la glicina por la arginina (Bellus et al. 1995, Shiang et al. 1994, Rousseau et al. 1996). El otro 1% de los casos (restantes) son causados por un punto mutado G a uno C dentro del nucleótido 1138. El gen mutantefue descubierto por John Wasmuth y sus colegas en 1994. Existen dos síndromes que tienen una base genética similar a la acondroplasia: hipocrondroplasia y la displasia tanatofórica.
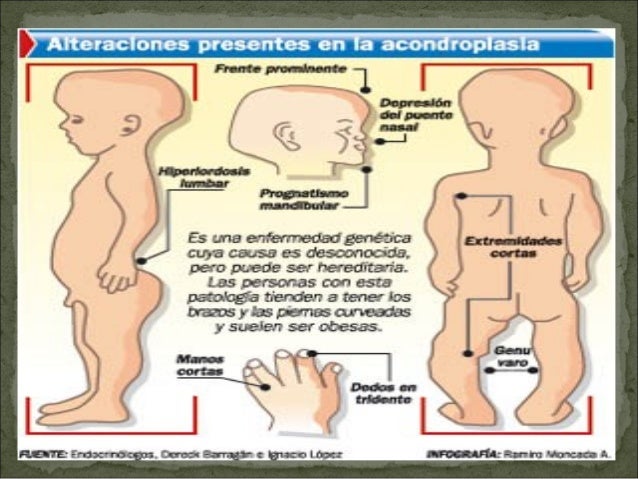
Signos
y síntomas clínicos
Signos
y síntomas clínicos
Diagnóstico
La acondroplasia puede ser detectada antes del nacimiento mediante un ultrasonido, el diagnóstico consiste en una ecografía fetal por discordancia progresiva entre la longitud del fémur y el diámetro biparietal por edad. Una prueba de ADN puede ser realizada antes del nacimiento para detectar la homocigosidad de la mutación, una condición que como ya se mencionó resulta letal
En la actualidad se ha localizado el gen transmisor en el
cromosoma 4.P 16.3 (brazo corto del cromosoma 4).
La expectativa de vida y el coeficiente intelectual de las
personas con acondroplasia son los mismos que los de las personas de talla
normal, a pesar de que los niños con este problema suelen tener un
desenvolvimiento motor lento cuando son bebés, a causa de las proporciones de
su cuerpo. Hay una serie de problemas derivados de la acondroplasia, a lo largo
de la vida, como otitis frecuentes, problemas con la columna, torcedura de
piernas, apneas en ocasiones, etc, pero la calidad de vida de los niños puede
mejorar con un seguimiento médico adecuado.
Desde un punto de vista social, las personas con
acondroplasia conviven siempre con una serie de problemas debidos a la
existencia en nuestra sociedad de clichés y prejuicios culturales e históricos
que aún persisten hoy en día. Un halo cómico o farandulesco rodea a las
personas acondroplásicas, que puede ser doloroso y traer consigo problemas
vitales muy serios para todas ellas.
La acondroplasia aparece como una mutación espontánea, que
tiene lugar por azar cada veinte mil nacimientos aproximadamente. Alrededor del
noventa por ciento de los niños con acondroplasia no tienen historia de ella en
sus familias.
La acondroplasia es causada, en el 97% de los casos, por una
mutación puntual debida a la sustitución de la Glicina 380 por Arginina en el
fragmento transmembranal del receptor 3 del factor de crecimiento fibroblástico
(FGFR3), aún cuando una mutación menos frecuente que también causa la
acondroplasia es la sustitución de la Glicina 375 por Cisteína.
FGFR3 pertenece a una familia de receptores estructuralmente
relacionados de quinasas dependientes de tirosina, y codificadas por cuatro
genes diferentes que originan múltiples variantes del receptor. Las mutaciones
en acondroplasia inducen activación excesiva del fragmento catalítico del
receptor, la quinasa dependiente de tirosina. El resultado es una elevada
actividad en las señales producidas por el receptor, originando una placa de
crecimiento defectuosa, en la cual las células no mantienen su patrón organizado
y, finalmente, no completan el proceso de diferenciación, lo que causa un
bloqueo en el crecimiento de los huesos.
La restauración del crecimiento normal en acondroplasia
podría, por lo tanto, obtenerse mediante la regulación específica de las señales
inducidas por el receptor en las células específicas dentro de la placa de
crecimiento, permitiendo, de tal manera, una ordenada y sincronizada elongación
ósea. Hasta el momento, no existe ningún tratamiento farmacológico para la
acondroplasia.
¿Qué es la Acondroplasia?

La acondroplasia es la forma más frecuente de enanismo. Se
trata de una alteración ósea de origen cromosómico, caracterizada porque todos
los huesos largos están acortados simétricamente, siendo normal la longitud de
la columna vertebral, lo que provoca un crecimiento disarmónico del cuerpo.
La acondroplasia es debida a un cambio en la información
genética que recibe el factor receptor de crecimiento de fibroplastos, células
que hacen que los huesos crezcan a lo largo. Esto produce una malformación en
el desarrollo de los cartílagos, con una calcificación acelerada que impide el
crecimiento normal de los huesos. Las personas con acondroplasia tienen un
torso de medida normal, las extremidades cortas y la cabeza ligeramente más
grande, además de otras características fenotípicas más o menos regulares.
Existe un elevado riesgo de muerte en la infancia debido a
la comprensión de la médula espinal y obstrucción de las vías respiratorias.
Los casos homocigóticos de acondroplasia son generalmente letales en el período
neonatal y llegan a afectar al 25% de la progenie de parejas en donde los dos
padres son heterocigotos para la enfermedad.
Existe relación en muchos de los casos entre la edad paterna
en el tiempo de la concepción de los individuos afectados y la acondroplasia.
Si una persona acondroplásica se une con una persona normal,
la probabilidad de que el hijo sea también acondroplásico es del 50%, cifra que
sube al 75% si ambos padres lo son (en este caso, hay un riesgo del 2,5% de
aparición de la acondroplasia homocigota, que suele ser letal al nacimiento).
Suscribirse a:
Comentarios (Atom)